Annotate single TCRs in the command line with autoDCR cli
Often when performing different TCR related analyses I’ve noticed that I need to quickly analyse one or two sequences, for which standard FASTA/FASTQ annotation pipelines (like autoDCR annotate, covered in the FASTA/Q V/J/CDR3 annotation with autoDCR annotate section) can be unwieldy. Therefore autoDCR has a command line interface option, which effectively allows you to just do a quick check of a potential rearranged TCR sequence. It is run using the autoDCR cli subcommand.
Note that by default autoDCR cli attempts to search in ‘full’ mode, rather than the ‘vjcdr3’ mode that is default for the annotate subcommand: see the discussion of the dcr_mode parameter in the Analysing full TCR transcripts section.
autoDCR cli [some-TCR-sequence] [-X -Y -Z other parameters]
# E.g. let's test a few examples using a long read TCR
# Note that I'm using a bash variable to hold it here for readability
# Pasting the TCR in directly also works
tcr="TACCGGCTATCTGTACGGGGACAGATACAGAAGACCCCTCCGTCATGCAGCATCTGCCATGAGCATCGGCCTCCTGTGCTGTGCAGCCTTGTCTCTCCTGTGGGCAGGTCCAGTGAATGCTGGTGTCACTCAGACCCCAAAATTCCAGGTCCTGAAGACAGGACAGAGCATGACACTGCAGTGTGCCCAGGATATGAACCATGAATACATGTCCTGGTATCGACAAGACCCAGGCATGGGGCTGAGGCTGATTCATTACTCAGTTGGTGCTGGTATCACTGACCAAGGAGAAGTCCCCAATGGCTACAATGTCTCCAGATCAACCACAGAGGATTTCCCGCTCAGGCTGCTGTCGGCTGCTCCCTCCCAGACATCTGTGTACTTCTGTGCCAGCAGACTGGACAGGGAGTACGAGCAGTACTTCGGGCCGGGCACCAGGCTCACGGTCACAGAGGACCTGAAAAACGTGTTCCCCTCT"
# Use the default 'full' autoDCR mode to analyse a long TCR read
autoDCR cli $tcr
TCR regions detected!
orientation forward
productive yes
v_call TRBV6-5*01
j_call TRBJ2-7*01
junction_aa CASRLDREYEQYF
l_call TRBV6-5*01
c_call TRBC2*01,TRBC2*02,TRBC2*03
# Analyse the same TCR sequence using the regular 'vjcdr3' mode
autoDCR cli $tcr -m vjcdr3
TCR regions detected!
orientation forward
productive yes
v_call TRBV6-5*01
j_call TRBJ2-7*01
junction_aa CASRLDREYEQYF
CLI output modes
autoDCR cli has some additional ways to report the annotations of the provided read, beyond just printing to the terminal, specified via the --output_mode / -om flag (default = stdout).
JSON output
The most information-rich option is to use -om json, which saves the entirety of the dictionary used internally by autoDCR for assessing each sequence, including all TCR tag matches and inferred properties, which may be more information than is useful for many applications.
autoDCR cli $tcr -om json -n my-cool-tcr
GenBank output modes
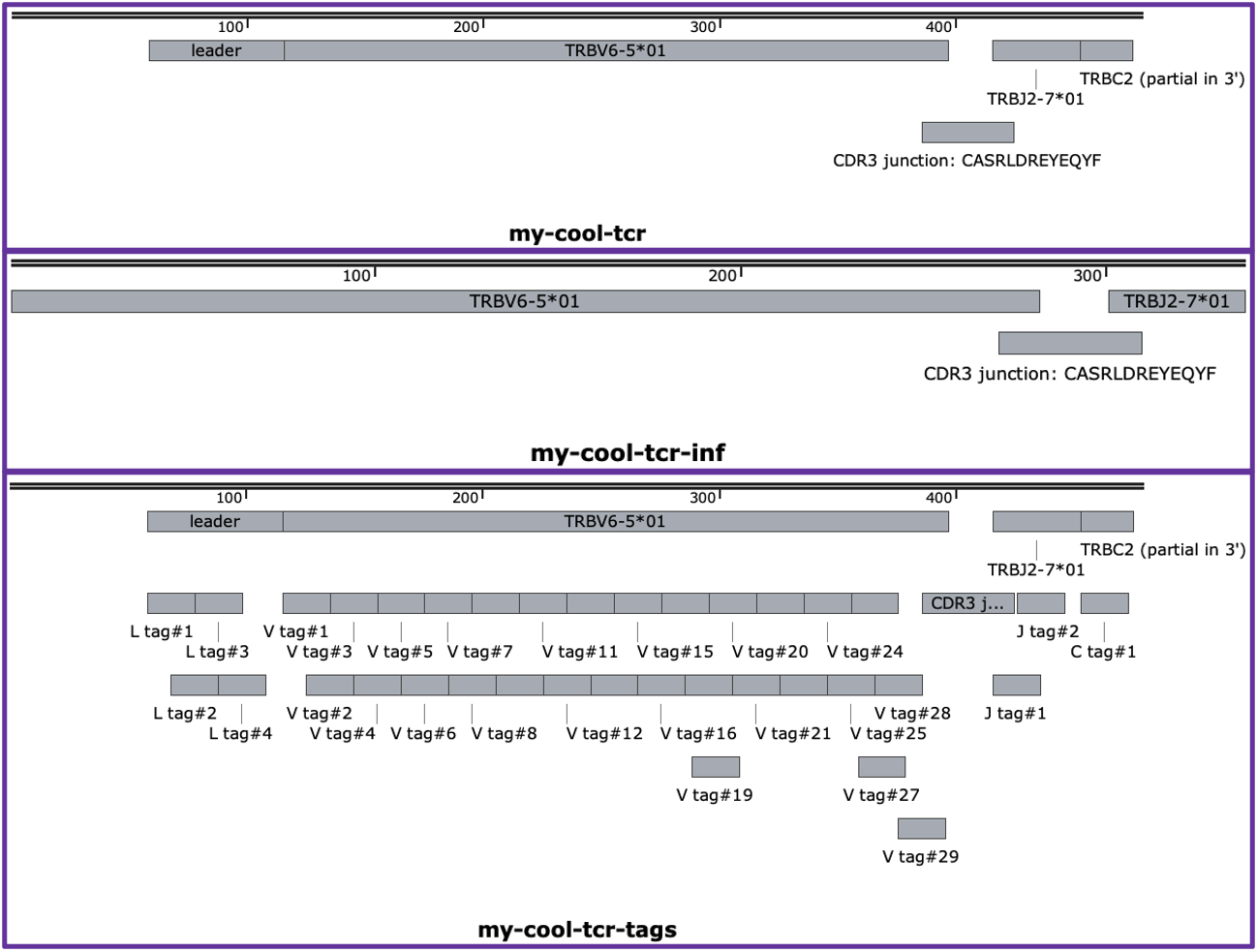
More useful to the average user is the ability to save a GenBank formatted summary file (produced using the functions developed for stitchr), which can then be viewed with a standard DNA browsing tool like SnapGene Viewer or ApE. See the following examples which use the --genbank_mode / -gm flag to specify different ways to generate a GenBank file (which also uses the --tcr_name / -n flag to provide output file names):
autoDCR cli $tcr -om json -n my-cool-tcr
autoDCR cli $tcr -om gb -n my-cool-tcr-inf -gm inferred
autoDCR cli $tcr -om gb -n my-cool-tcr-tags -gm tags
Then compare the output of the GenBank files produced in the image below, screenshot from SnapGene Viewer.
The first option uses the default ‘read’ output mode, which just annotates the read as provided with the detected regions.
The second ‘inferred’ option instead annotated the ‘full inferred’ TCR produced by
autoDCRduring its analysis (which is more useful in shorter read TCR sequences).The final option shows the results of ‘tags’, in which the individual location of matched tags are recorded, alongside the full regions. This can be particularly useful for investigating strange TCR reads (and is used a lot when developing novel
autoDCRfunctions).
Things to note
When analysing a single TCR, and using an output mode that produces a file, providing a name via the
--tcr_name / -nflag is not required. However if you don’t use itautoDCRwill automatically generate a file name based on the rearrangement detected. E.g. the products of the example TCR read above would be titledautoDCR-TCR_TRBV6-5-01|V_TRBJ2-7-01|J_CASRLDREYEQYF.[suffix]Given their length (and frequently being sequenced at the start of an Illumina read 2, with correspondingly lower read quality) leader regions are often not able to be unambiguously determined. Similarly many molecular protocols reverse transcribe and/or amplify from the 5’ end of the constant region, and thus almost none of the allele-distinguishing polymorphisms are sequenced. Such regions will therefore (where possible) be reported at the gene level, or just as ‘leader’ in output GenBank files. However if an unexpected leader is detected (i.e. not the sequence associated with that V-REGION) it will be labelled in full.